pPOD Project: Helping Assemble the Tree of Life
 The pPOD team at UC Davis recently announced a preview release of a Kepler extension that provides new actors and tools to create and manage phylogenetic analyses. This extension was developed as part of a 3-year National Science Foundation grant to address informatics challenges faced by researchers funded by the AToL (Assembling the Tree of Life) initiative. The pPOD extension includes actors that will enable AToL teams to automate phylogenetic analyses as well as reliably record and later reconstruct how results were obtained from primary observations.
The pPOD team at UC Davis recently announced a preview release of a Kepler extension that provides new actors and tools to create and manage phylogenetic analyses. This extension was developed as part of a 3-year National Science Foundation grant to address informatics challenges faced by researchers funded by the AToL (Assembling the Tree of Life) initiative. The pPOD extension includes actors that will enable AToL teams to automate phylogenetic analyses as well as reliably record and later reconstruct how results were obtained from primary observations.
The pPOD extension, bundles the Kepler beta release with new pPOD actors, a number of phylogenetics applications automated by the actors, and sample workflows. The sample workflows highlight how Kepler can be used to align nucleotide and protein sequences, and to infer phylogenetic trees from these alignments using maximum likelihood, maximum parsimony, and Bayesian methods implemented in widely used tools. The workflows also demonstrate how easily native Java actors and ones that automate external applications or employ remote services can interoperate in a single Kepler workflow. A number of the actors in the package use the Gblocks application and programs in the Phylip application suite, all of which are bundled in the release. Other actors employ the REST services provided by the CIPRES Portal hosted at the San Diego Supercomputer Center (SDSC), to transparently run the PAUP*, RAxML, MrBayes, and CLUSTAL applications on SDSC computer systems.
|
|
|
Example of a Kepler workflow that uses the COMAD computational model, displayed with Workflow trace as displayed by the new provenance browser bundled with the pPOD preview release. |
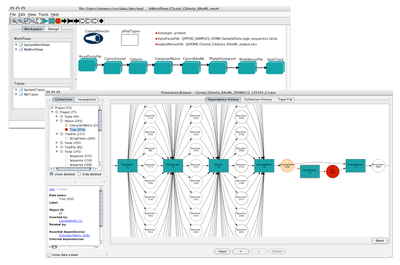
The pPOD actors and workflows are based on the Collection-Oriented Modeling and Design (COMAD) paradigm, a new computational model featuring built-in support for processing nested data collections in an assembly-line manner, and a fine-grained method for capturing and representing data provenance. Because of the often nested structure of biomolecular data sets, COMAD is well-suited for automating phylogenetics and other bioinformatics workflows. COMAD also facilitates the development of custom data types for particular domains. The pPOD package includes a custom data model for phylogenetics that allows actors that wrap applications and services employing different data formats to be strung together without intervening format-conversion actors or shims. Data is transparently provided to, and retrieved from, underlying software applications using this data model so that workflows focus on the scientific rather than the data-manipulation aspects of computations. Consequently, the scientific intent of these analyses can be read directly from the Workflow canvas (see Figure).
pPOD workflows also exploit the provenance capabilities provided by COMAD. Each run of the sample pPOD workflows produces an execution trace file. A trace is an XML representation of the data and collections input to and created by a workflow run, the parameter values for the actors, and the detailed provenance of all data created during the run. These traces are listed in a new panel within the Kepler GUI and can be viewed in an interactive provenance browser application bundled with the pPOD preview release (see Figure). All source code developed by the pPOD project for Kepler, including the provenance browser application, are available via the Kepler source code repository. The Kepler/pPOD preview distribution for OS X, along with additional information about the project, can be found at http://daks.genomecenter.ucdavis.edu/kepler-ppod/.
